

Zusammenfassung
Ichthyosen sind genetisch bedingte Verhornungsstörungen der Haut, die durch eine starke Schuppung gekennzeichnet sind. Man unterscheidet zwischen Formen, die nur die Haut und solchen, die auch andere Organe betreffen. Als Ursache der Ichthyosen wird eine gestörte Hautbarriere vermutet, die als Reparaturversuch eine verstärkte Hornhautbildung auslöst. Nach heutigem Stand ist weder eine Heilung noch eine ursächliche Therapie möglich. Zur Symptomverbesserung ist eine intensive, tägliche und lebenslange Hautpflege notwendig.
Auf einen Blick
+ Auftreten Entweder von Geburt an (kongenital) oder in Laufe der Kindheit auftretend
+ Symptome Verstärkte Hornhautbildung, sichtbare graue bis braune Schuppung, teilweise starke Entzündung der Haut, je nach Form weitere Symptome und Behinderungen möglich
+ Einflussfaktoren Genetisch bedingt. Stark gestörte Hautbarriere; trockene Luft, Kälte verschlimmern Symptomatik
+ Ansteckungsgefahr Keine – Gendefekt vererbbar

Einführung
Unter dem Begriff „Ichthyosen“ (griech. „ichthys“ = Fisch) werden verschiedene genetisch bedingte Verhornungsstörungen der Haut zusammengefasst, die mit einer vermehrten Hornhautbildung und einer dadurch ausgelösten, starken Schuppung meist des gesamten Körpers einhergehen. Umgangssprachlich bezeichnet man diese Krankheit aufgrund ihrer Erscheinungsform auch als Fischschuppenkrankheit. Allen Krankheitsformen innerhalb dieser heterogenen Gruppe ist gemein, dass sie durch Genveränderungen (Mutationen) entstehen, die den Funktionsausfall von Eiweißen auslösen, die für eine stabile Hautbarriere benötigt werden. Die vermehrte Hornhautbildung wird heute als ein Versuch des Körpers gesehen, die gestörte Hautbarriere zu reparieren. Früher wurden die Ichthyosen in angeborene Formen und solche eingeteilt, die sich erst im Laufe des Lebens ausprägen. Heute unterscheidet man dagegen zwischen Formen, bei denen ausschließlich die Haut betroffen ist und solchen, bei denen die Ichthyose nur Teil eines übergeordneten Syndroms ist, so dass auch andere Organe beeinträchtig sind.
Zu den Ichthyosen gehören vergleichsweise häufige Formen wie die Ichthyosis vulgaris mit einer Erkrankungshäufigkeit von 1:100 (ein Erkrankter pro hundert Menschen), aber auch sehr seltene Formen. Ichthyosen, die über das weibliche Geschlechtschromosom (X-Chromosom) übertragen werden, betreffen stärker oder ausschließlich Jungen und Männer, da Frauen einen Gendefekt auf einem X-Chromosom durch die gesunde Genkopie auf dem zweiten X-Chromosom ausgleichen können. Schwere Formen wie die meisten angeborenen Formen und vor allem die bereits während der Embryonalentwicklung auftretende Harlekin-Ichthyose können für Säuglinge lebensbedrohlich sein, da es durch die gestörte Hautbarriere zu einem großen Wasserverlust kommt und auch die Wärmeregulationsfunktion der Haut gestört ist. Zudem treten als Folge der Erkrankung häufig bakterielle Hautinfektionen auf. Die den Ichthyosen zugrunde liegenden Gendefekte, die heute entschlüsselt sind, lassen sich weder ursächlich therapieren, noch ist eine Heilung möglich. Für die Betroffenen ist eine daher intensive und regelmäßige Hautpflege essentiell, um die Hautbarriere zu stärken und bakterielle Infektionen der geschädigten Haut zu verhindern. Dazu werden die Hornschuppen mechanisch und/oder mit äußerlich anwendbaren (topischen) Wirkstoffen entfernt. Anschließend ist eine effektive Rückfettung der Haut unerlässlich.
Ursachen und Auslöser
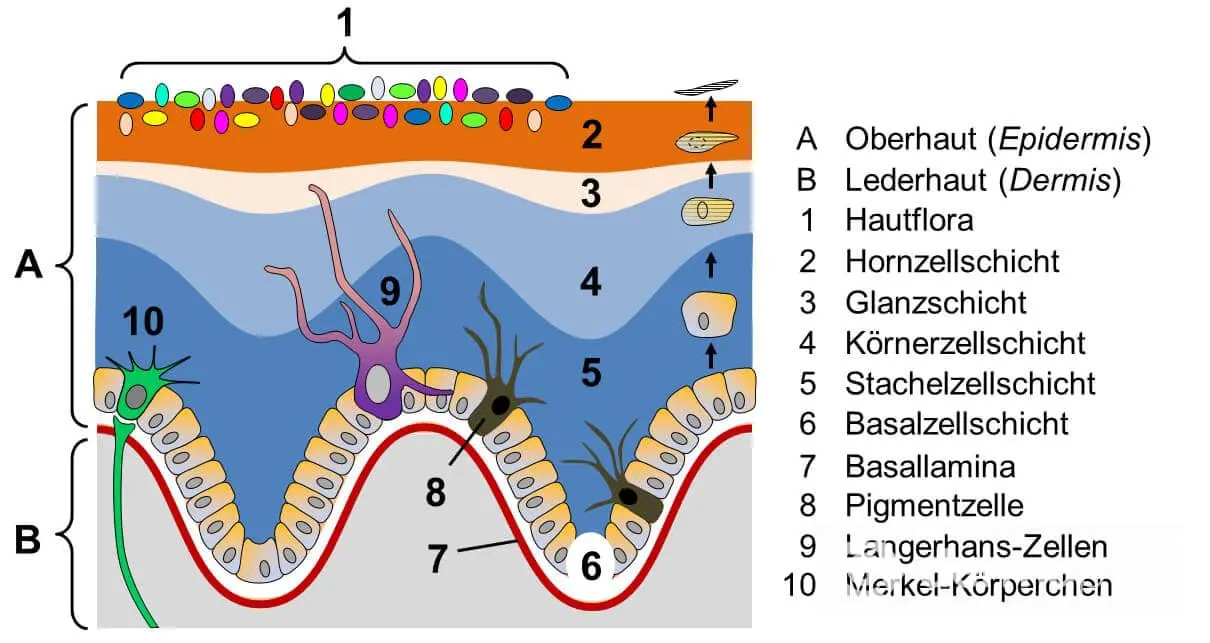
Die äußere Schicht der Haut, die Oberhaut (Epidermis, Abb. 1: A), dient als Barriere gegenüber Umwelteinflüssen. Dabei dient die Hautbarriere nicht nur dazu, das Eindringen von giftigen Substanzen und Krankheitserregern in tiefere Hautschichten, sondern auch den Wasserverlust über die Haut zu verhindern. Eine Gemeinsamkeit aller Ichthyosen ist das Fehlen bestimmter Eiweiße, die zur Bildung der Hautbarriere beitragen. Um die Defekte in der Hautbarriere auszugleichen, produziert der Körper vermehrt Zellen der Oberhaut, die verhornen und als Hornschuppen abschilfern.
Autosomal-dominante Ichthyosis vulgaris
Bei der Ichthyosis vulgaris, der häufigsten Form der Ichthyosen und einer der häufigsten erblichen Erkrankungen überhaupt, ist das Eiweiß Filaggrin betroffen. Ursächlich ist hier ein autosomal-dominant vererbter Gendefekt. Ein Drittel der Betroffenen weist nur eine einzige Veränderung im Filaggrin-Gen auf, bei den restlichen zwei Dritteln sind es zwei Veränderungen, wodurch das Filaggrin noch stärker beeinträchtigt wird. Durch das Fehlen des funktionellen Proteins kommt es zu einem vermehrten Wasserverlust über die Epidermis und einer geringeren Feuchtigkeit der Hornschicht (Stratum corneum, Abb. 1: 2), der äußersten Schicht der Oberhaut.
X-chromosomal-rezessive Ichthyosis
Die X-chromosomal-rezessive Ichthyosis tritt nur beim männlichen Geschlecht auf, da Frauen den Defekt mit einer intakten Genkopie auf dem zweiten X-Chromosom ausgleichen können. Allerdings können solche „stillen Defektträger“-Frauen das defekte Gen an ihre Nachkommen weitergeben. Bei der X-chromosomal rezessiven Ichthyose ist das Enzym Steroidsulfatase beeinträchtigt, das in 90% der Fälle vollständig fehlt. Dadurch kommt es zu einer Anhäufung des Stoffwechselprodukts Cholesterolsulfat, das bestimmte proteinabbauende Enzyme in der Oberhaut hemmt und so die Abschuppung der Hornzellen verhindert.
Je nachdem, ob auch weitere angrenzende Bereiche des Chromosoms fehlen, können Krankheitsformen auftreten, die andere Organe in Mitleidenschaft ziehen und begleitende Symptome hervorrufen. So zeigen 40% der Patienten zusätzlich ein Aufmerksamkeitsdefizit-Hyperaktivitätssyndrom (ADHS), 25% eine Form von Autismus und 20% einen Hodenhochstand (Kryptorchismus). Mit einer Erkrankungshäufigkeit (Prävalenz) von 1:4000 gehört die X-chromosomal rezessive Form der Ichthyose zu den seltenen Krankheiten.
Refsum-Syndrom
Eine spezielle Form der Ichthyose ist das Refsum-Syndrom, bei dem es zu einer Anhäufung von Phytansäure im Gewebe und Blut kommt. Diese Fettsäure wird mit der Nahrung aufgenommen und kann von den Patienten entweder nicht abgebaut oder nicht in die Zellen aufgenommen werden, so dass sie sich im Gewebe anhäuft. Ohne Behandlung verläuft diese Erkrankung nach dem Auftreten im frühen Erwachsenenalter fortschreitend und geht neben der Ichthyose vor allem mit neurologischen Symptomen einher. Eine Diät, die arm an Phytan ist, kann jedoch den fatalen Verlauf verhindern.
Autosomal-rezessive kongenitale Ichthyosen (ARCI)
Unter dem Sammelbegriff „Autosomal-rezessive kongenitale Ichthyosen“ (ARCI) fasst man sämtliche Formen zusammen, die neben der Haut weitere Organsysteme betreffen, sich von Geburt an manifestieren (kongenital), keine Tendenz zur Blasenbildung der Haut zeigen und nicht über die Geschlechtschromosomen (autosomal) vererbt werden.
Die Krankheit entsteht nur, wenn beide, die mütterliche und die väterliche, Genkopie betroffen sind (rezessive Vererbung). Deshalb sind diese Formen sehr selten. Zu den ARCI gehören verschiedene Ausprägungen der lamellären Ichthyosis, der ichthyosiformen Erythrodermie (Erythodermie = Rötung des gesamten Hautorgans) und die Harlekin-Ichthyose, die schwerste bekannte Form, die sich bereits im Mutterleib ausprägt und für den Fötus und das Neugeborene tödlich sein kann. Die Gendefekte dieser Formen haben Auswirkungen auf unterschiedliche Eiweiße:
Ein Mangel des Enzyms Transglutaminase-1 macht in Deutschland 30% der ARCI-Fälle aus. Es handelt sich dabei mit einer Prävalenz von 1:200.000 um den häufigsten Defekt innerhalb des ARCI-Spektrums. Zusammen 10% entfallen auf Defekte in zwei verschiedenen Genen für das Enzym Lipoxygenase. Sowohl die Transglutaminase-1 als auch die Lipoxygenase sind an der Ausbildung der Protein- und Lipidhülle der Hornzellen (Korneozyten) der Epidermis beteiligt.
Dagegen gibt es verschiedene andere Gendefekte, die noch seltener sind, wie solche im Gen des Lipidtransporters ABCA12. Ein Totalausfall des Transporters führt zu einem gravierenden Defekt des Lipidaufbaus in der Hornschicht der Oberhaut und zu der schwersten bekannten Form der Ichthyose (Harlekin-Ichthyose).
Keratinopathische Ichthyosen
Ichthyosen, die auf eine Veränderung der Hornsubstanz, dem Eiweiß Keratin, zurückgehen, werden unter dem Begriff „keratinopathische Ichthyosen“ zusammengefasst. Keratine sind Strukturproteine, und durch ihren Funktionsverlust verlieren die Zellen der Oberhaut ihren Zusammenhalt, wodurch Hautblasen entstehen. Je nachdem, welches der verschiedenen Keratingene verändert ist, variieren Krankheitsbild und Schweregrad, und es können unterschiedliche Schichten der Oberhaut betroffen sein. Eine Form, bei der zur Ichthyose verschiedene andere schwere Symptome und Behinderungen hinzukommen, ist die kongenitale retikuläre ichthyosiforme Erythrodermie (KRIE), auch als Konfetti-Ichthyosis bekannt, da sich auf der Haut im Kindesalter weiße Inseln herausbilden.
Symptome und Krankheitsverlauf
Auffälligstes Symptom der Ichthyosen ist die starke Schuppung der Oberhaut, die bei allen Formen auftritt und meist den gesamten Körper betrifft. Der Schweregrad, der Verlauf und hinzukommende Symptome unterscheiden sich dagegen bei den einzelnen Formen. Häufig, vor allem bei angeborenen Ichthyosen, kommt es auch zu einer ausgeprägten Entzündungsreaktion der Haut.
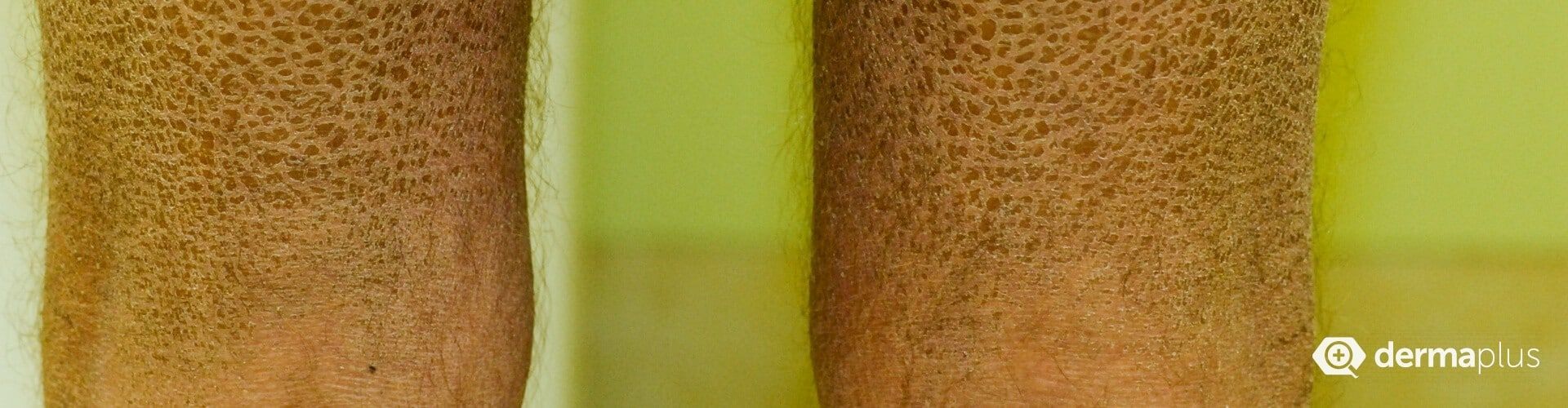
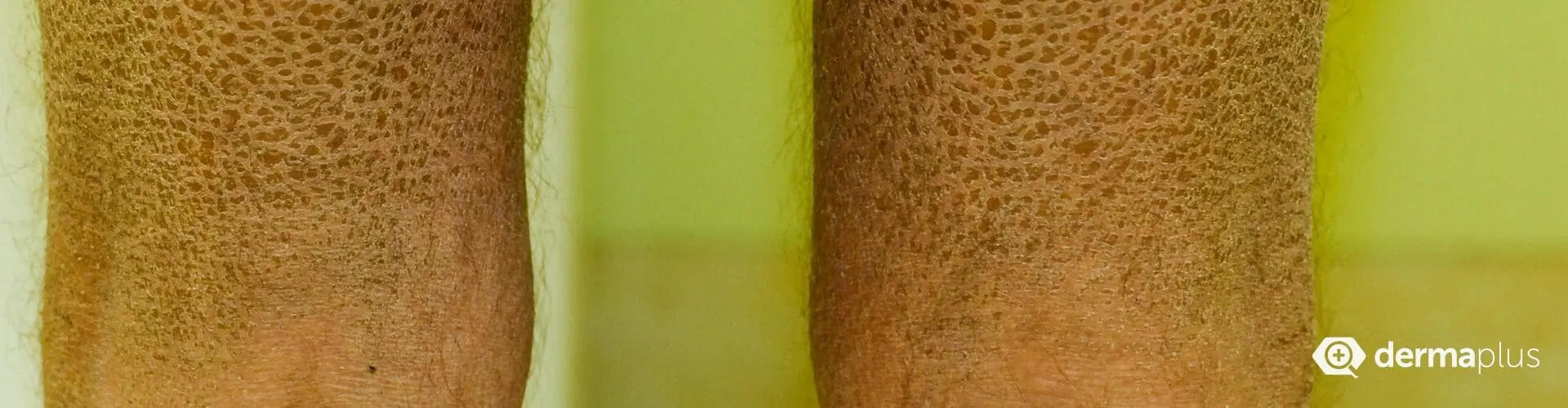
Patienten mit Ichthyosis vulgaris, der häufigsten Form der Ichthyosen, weisen eine Orthohyperkeratose auf, bei der sich die hornbildenden Zellen der Oberhaut zwar normal entwickeln, sich aber zu stark vermehren, sodass sich eine verdickte Hornschicht bildet (Orthohyperkeratose). Typisch im Erscheinungsbild sind feine, meist hellgraue Schuppen aus abgestorbener Haut, die das Gesicht und die Gelenkbeugen aussparen (Abb. 3 und 4). Die Symptome beginnen meist im Säuglingsalter (Abb. 2) und bessern sich im Sommer bei höheren Temperaturen. Auch eine höhere Luftfeuchtigkeit wirkt sich günstig aus. Da Filaggrin-Defekte, die für die Entstehung der Ichthyosis vulgaris verantwortlich sind, auch bei der Entstehung der atopischen Dermatitis (Neurodermitis) eine Rolle spielen, entwickelt etwa die Hälfte der Ichthyosis vulgaris-Patienten zusätzlich diese oder eine andere atopische Erkrankung wie Heuschnupfen oder allergisches Asthma. Viele Patienten können nur schlecht schwitzen und sind deshalb hitzeempfindlich. Außerdem weisen sie oft vertiefte Linien (Hyperlinearität) auf den Handflächen (Abb. 5) und Fußsohlen auf.
Patienten mit der X-chromosomal rezessiv vererbten Ichthyose bilden im Unterschied zur Ichthyosis vulgaris rhombische (rautenförmige), festhaftende Schuppen, die den gesamten Körper bedecken und Ellenbeugen, Kniekehlen, Handteller sowie Fußsohlen aussparen. Die Hyperlinearität fehlt und die Schuppen sind eher bräunlich als grau. Weitere Symptome wie eine Hornhauttrübung, Hodenhochstand, ADHS und Autismus können hinzukommen und Mütter erkrankter Kinder berichten oft über Geburtskomplikationen. Wenn größere Abschnitte des entsprechenden Chromosoms fehlen, können übergeordnete Syndrome entstehen wie die Chondrodysplasia punctata, die durch Skelett- und Knorpelfehlbildungen gekennzeichnet ist.
Die Erscheinungsform der ARCI ist extrem variabel, da verschiedene Gene betroffen sein können. Typisch sind eine verbreiterte Epidermis, die durch verstärktes Wachstum der Oberhaut zustande kommt. Die Verhornung ist wie bei der Ichthyosis vulgaris verstärkt, aber in ihrem Ablauf nicht gestört. Zusätzlich laufen in der Lederhaut (Dermis) oft starke entzündliche Prozesse ab. Ein Mangel an Transglutaminase-1 zeigt sich ebenfalls durch die Bildung von Spalten in der Hornschicht der Oberhaut, während Mutationen in den Genen des Lipidtransporters ABCA und in der Lipoxygenase ALOX veränderte „lamellar bodies“ zur Folge haben. Diese Körperchen sind am Transport von Fetten beteiligt und werden für die Aufrechterhaltung der Hautbarriere benötigt. Die meisten Fälle von ARCI zeigen sich bereits bei der Geburt: Betroffene Neugeborene sind von einer glänzenden Kolloidmembran umgeben, die sich innerhalb der ersten vier Lebenswochen ablöst. Dieses typische Erkrankungsbild hat den Begriff „Kollodiumbaby“ geprägt. Manche Kinder sind anschließend vollständig geheilt, ein Phänomen, das als „Selbstheilendes Kollodiumbaby“ bezeichnet wird. Meist entwickelt sich jedoch eine klassische, schwere kongenitale Ichthyose mit dunkelbraunen, lamellären Schuppen, die typischerweise auch die großen Gelenkbeugen nicht aussparen und mit leichten Entzündungsreaktionen einhergehen. Manchmal steht dagegen die Entzündung im Vordergrund und die Schuppung ist eher fein und hellgrau. Auch eine Verhornung der Handflächen und Fußsohlen (palmoplantare Hyperkeratosen) kann auftreten. Bei der Badeanzug-Ichthyose bilden sich die Schuppen selektiv an Armen und Beinen zurück.
Eine besonders schwere Form ist die Harlekin-Ichthyose, bei der die Kinder oft bereits im Mutterleib versterben oder aber mit einem dicken und starren Hautpanzer geboren werden. Dieser schränkt nicht nur die Bewegung, sondern auch die Atmung und die Nahrungsaufnahme massiv ein. Zusätzlich zeigen die Kinder eine Auswärtswendung der Augenlider und der Lippen sowie eine Unterentwicklung von Ohr- und Nasenknorpel. Die geschädigte Haut neigt zum Aufreißen und ist extrem infektionsanfällig, sodass rund die Hälfte der erkrankten Säuglinge auch heute noch an der Erkrankung und ihren Folgen stirbt. Aufgrund der massiven Störung der Hautbarriere ist eine Austrocknung und Unterkühlung der Säuglinge häufig, sodass diese intensivmedizinisch betreut werden müssen. Überlebende Kinder zeigen ausgeprägte, lamelläre Schuppen und starke Entzündungsprozesse mit einhergehender Rötung der Haut (Erythrodermie). Später führt eine fehlende Fähigkeit zu Schwitzen meist zu einer verminderten Hitzetoleranz.
Bei den keratinopathischen Ichthyosen kommt es zu einer epidermolytischen (= mit Lösung der Epidermis einhergehenden) Hyperkeratose. Dabei bilden sich Hautblasen, da die hornbildenden Zellen (Keratinozyten) der Oberhaut aufgrund des defekten Keratins ihren Zusammenhalt verloren haben. Im Laufe der ersten Lebensmonate geht die Blasenbildung zurück und dafür entstehen ausgeprägte, stachelartige Schuppen, vor allem in Bereichen mit höherer Körpertemperatur wie den Achselhöhlen. Auch Handteller und Fußsohlen können betroffen sein.
Patienten mit der speziellen Form der Konfetti-Ichthyose sind oft insgesamt schwerkrank und weisen als Säugling in der Regel eine verzögerte Entwicklung auf. Auf Grund einer Spontanheilung in einzelnen Hautarealen entstehen bis zu 2 cm große, weiße Flecken ohne Schuppenbildung.
Erschwerend leiden Patienten mit schweren Formen der Ichthyose an einem Vitamin D-Mangel und entsprechenden Folgeproblemen, da der Haut in der Vitamin D-Produktion eine wichtige Funktion zukommt.
Diagnose und Differentialdiagnose
Die Diagnosestellung beginnt mit einer gründlichen Eigenanamnese, in der die Krankengeschichte des Patienten aufgenommen wird. Da es sich bei den Ichthyosen um eine Geno-Dermatose handelt, also eine genetisch bedingte und damit vererbbare Erkrankung der Haut, wird zusätzlich eine Familienanamnese durchgeführt und das Auftreten entsprechender Erkrankungen in der Verwandtschaft erfragt. Die verschiedenen Ichthyoseformen können meist anhand des Krankheitsverlaufs, Zeitpunkt des Auftretens und der Erscheinungsform (Blasenbildung, Schuppenform, Hautrötung) voneinander unterschieden werden. Dazu werden Haut, Schleimhäute und je nach Krankheitsbild auch die Hautanhangsorgane wie Haare und Nägel zunächst durch den Hautarzt untersucht. Im Zweifelsfall kann die histologische oder elektronenmikroskopische Untersuchung einer Hautbiopsie aufschlussreich sein. Je nach Ichthyoseform kann die Struktur der Oberhaut variieren, was sich histologisch im mikroskopischen Bild nachweisen lässt. So fehlt beispielsweise bei der Ichthyosis vulgaris die Körnerzellschicht (Stratum granulosum, Abb. 1: 4) der Oberhaut, während diese bei der X-chromosomal rezessiven Ichthyosis verbreitert ist. In Einzelfällen kann eine Bestimmung der Enzymaktivtät zeigen, ob ein Enzym wie beispielsweise die Transglutaminase-1 einen Defekt aufweist.
Bildet die Haut Blasen, sollten differentialdiagnostisch verschiedene Krankheiten ausgeschlossen werden. Dazu zählen u. a. die erblich bedingte Epidermolysis bullosa (Schmetterlingskrankheit), eine angeborene Zinkaufnahmestörung (Acrodermatitis enteropathica), erblich bedingte Abbaudefekte des Blutfarbstoffs Hämoglobin (Porphyrien), verschiedene blasenbildende Autoimmunerkrankungen sowie die bakterielle Infektionskrankheit Impetigo infectiosa (Grind- oder Borkenflechte). Der Ausschluss erfolgt durch verschiedene immunologische, mikrobiologische oder histochemische Laboruntersuchungen einer Hautbiopsie.
Eine relativ neue Möglichkeit zur zweifelsfreien Diagnose genetisch bedingter Erkrankungen ist der molekularbiologische Nachweis der Genveränderung. Vor allem bei Neugeborenen bietet die Molekulargenetik oft die einzige sinnvolle diagnostische Möglichkeit. Dabei können mehrere Kandidatengene gleichzeitig untersucht werden, was die Belastung für den Patienten reduziert und Zeit spart. Ein weiterer Vorteil ist, dass die Diagnostik zum Teil bereits vor der Geburt im Rahmen einer Chorionzottenbiopsie (Plazenta-Punktion) oder einer Amniozentese (Fruchtwasseruntersuchung) durchgeführt werden kann.
Therapie und Behandlung
Aufgrund der vielen unterschiedlichen Erkrankungsformen ist für die Behandlung der Ichthyose eine genaue Diagnostik unerlässlich. Ichthyosen sind nicht heilbar und auch nicht ursächlich behandelbar. Stattdessen werden in erster Linie die Symptome gelindert und vor allem die Hautbarriere gestärkt. Dazu gehört eine Entfernung der störenden Hautschuppen, eine Rückfettung der Haut und das Verhindern bzw. Behandeln von bakteriellen Folgeinfektionen. In schweren Fällen ist die Gabe von systemischen Wirkstoffen zur Regulation des Verhornungsprozesses und eine lokale antientzündliche Therapie möglich.
Inzwischen gibt es erste Ansätze, die zugrundeliegenden Gendefekte der Ichthyosen im Rahmen einer Gentherapie zu heilen. Dazu wird eine korrekte Genvariante in körpereigene Zellen eingefügt, was bei der Haut vergleichsweise einfach ist, da sie an der Körperoberfläche liegt und so mit minimalinvasiven Methoden zugänglich ist. Gentherapien für die Behandlung von Ichthyosen sind aber noch rein experimentell, d.h. sie stehen noch nicht für Routinebehandlungen zur Verfügung. Ein anderer Ansatzpunkt ist die Beeinflussung der Enzymaktivitäten, entweder medikamentös, wenn noch eine Restaktivität des körpereigenen Enzyms vorhanden ist oder durch Zugabe des jeweiligen Enzyms von außen (Substitution).
Eine Voraussetzung für den therapeutischen Erfolg sind die Bereitschaft zur Mitarbeit und die Therapietreue der Patienten. Aufgrund der vielen unterschiedlichen Ichthyoseformen ist eine Beratung in einem spezialisierten, dermatologischen Zentrum zu empfehlen.
Fischschuppenkrankheit – Örtliche Therapien
Zur Verringerung der Schuppung wird gezielt die Feuchtigkeit der Haut erhöht. Hierzu stehen verschiedene Zubereitungen mit den Wirkstoffen Harnstoff, Milchsäure, Glyzerin, Dexpanthenol oder Macrogol 400 zur Verfügung. Vor allem Harnstoff stellt aufgrund seiner wasserbindenden, barriereregenerierenden, entschuppenden und antimikrobiellen Eigenschaften einen der wichtigsten Wirkstoffe dar. Allerdings wird er ebenso wie Milchsäure bei stark entzündlichen Ichthyoseformen oft schlecht toleriert. Die Verträglichkeit beider Wirkstoffe kann durch Zusatz von 20%igem Polyethylenglykol verbessert werden. Dennoch sollte im ersten Lebensjahr auf Harnstoff verzichtet werden, da es zu Hautreizungen und einem erhöhten Harnstoffspiegel im Blut kommen kann. Das Gleiche gilt für Salizylsäure, die in einer Konzentration von 10% hautschälende Wirkung hat, bei Säuglingen und Kleinkindern aber schnell schädliche Konzentrationen im Blut erreichen kann. Vitamin-A-Säure-haltige Zubereitungen sollten ebenfalls nur bei Erwachsenen zum Einsatz kommen sollten.
Bei leichteren Formen der Ichthyosis wie der Ichthyosis vulgaris und der X-chromosomalen rezessiven Ichthyosis reicht das zweimal tägliche Eincremen mit Zubereitungen aus, die die oben genannten Wirkstoffe enthalten. Patienten mit schweren Verlaufsformen der ACRI oder mit keratinopathischen Formen müssen zusätzlich ein tägliches Vollbad nehmen und danach die Schuppen mechanisch durch einen Schwamm, ein Mikrofasertuch oder Bimsstein entfernen. Anschließend erfolgt das Eincremen des gesamten Körpers bei noch nasser Haut mit der rückfettenden Basistherapie. Im Badewasser scheint ein Zusatz von Natriumbicarbonat (Backpulver) die Wasseraufnahme durch die Hornschicht zu verbessern.
Entzündungen der Haut können mit Calcineurininhibitoren wie Tacrolimus und Pimecrolimus bekämpft werden. Allerdings besteht aufgrund der gestörten Hautbarriere die Gefahr, dass zu viel des Wirkstoffs über die Haut aufgenommen wird. Deshalb sollte, wenn möglich, keine großflächige Anwendung erfolgen, und ansonsten die Wirkstoffkonzentration im Blut ärztlich kontrolliert werden.
Fischschuppenkrankheit – Systemische Therapien
Retinoide wie Acitretin und Etretinat, die strukturell dem Vitamin A verwandt sind, können auch systemisch angewendet werden. Sie modulieren das Wachstum und die Reifung der hornbildenden Zellen der Oberhaut und wurden deshalb bis vor kurzem auch für die Therapie der Schuppenflechte (Psoriasis) eingesetzt, einer entzündlichen Hautkrankheit, die ebenfalls mit einer vermehrten Bildung von Hautzellen einhergeht. Vitamin A ist ein Molekül, das in Entwicklungsvorgänge eingreift. Deshalb sollten auch seine Derivate nur bei Patienten eingesetzt werden, die die Wachstumsphase abgeschlossen haben. Retinoide sind potenziell leber- und fruchtschädigend, so dass bei Frauen im fruchtbaren Alter eine sichere Empfängnisverhütung erfolgen muss. Lediglich bei der Harlekin-Ichthyose ist die Gabe von systemischen Retinoiden aufgrund der schweren Verlaufsform auch schon im Neugeborenenalter zu rechtfertigen. Sie werden vor allem eingesetzt, um die Abstoßung des bei der Geburt vorhandenen Hautpanzers zu beschleunigen. Des Weiteren sollten Folgeerkankungen wie bakterielle Infektionen und Vitamin D-Mangel behandelt werden.
Behandlung des Kollodiumbabys
Neugeborene mit Harlekin-Ichthyose und Kollodiumbabys sollten immer als Notfall gelten und umgehend intensivmedizinisch behandelt werden. Da aufgrund der gestörten Hautbarriere ein großer Wasserverlust über die Haut erfolgt, werden die Neugeborenen in einem Inkubator mit hoher Luftfeuchte (70-80%) versorgt. Außerdem werden das Körpergewicht, der Elektrolyt- und der Wärmehaushalt überwacht. Bei Bedarf kann Flüssigkeit über einen Nabelvenenkatheter gegeben werden, was jedoch das Risiko einer bakteriellen Infektion birgt. Im ersten Lebensjahr wird die Haut mit rückfettenden Zubereitungen (ohne Wirkstoff) gepflegt. Für die Augen empfiehlt sich die Pflege mit einer speziellen Augensalbe wie dem dexpanthenolhaltigem Corneregel ®.
Selbsthilfegruppen
Hilfe finden Betroffene und ihre Angehörigen in der Patientenorganisation Selbsthilfe Ichthyosis e. V. (https://ichthyose.de/). Informationen über das Krankheitsbild erhält man auch auf der Webseite des Deutschen Ichthyosenetzwerkes NIRK (www.netzwerk-ichthyose.de). Bei Kinderwunsch kann eine genetische Beratung durch einen Humangenetiker sinnvoll sein.
Prävention und Vorbeugung
Da die Ichthyosen durch Gendefekte verursacht werden, ist eine Prävention nicht möglich.
Quellen und weiterführende Literatur
- Has C, He Y, 2016, Praktische Aspekte der molekularen Diagnostik bei Genodermatosen, Hautarzt 67: 53-58.
- Krug M et al., 2009, Ichthyosen – Teil 1: Differentialdiagnose vulgärer Ichthyosen und therapeutische Erwägungen, Journal of the German Society of Dermatology 7: 511-520.
- Krug M et al., 2009, Ichthyosen – Teil 2: Kongenitale Ichthyosen, Journal of the German Society of Dermatology 7: 577-588.
- Traupe H et al., 2014, Die nichtsyndromalen Ichthyosen – aktueller Stand, Journal of the German Society of Dermatology, DOI: 10.1111/ddg.12229.