Nicht infektiöse Granulome der Haut – Zurückhaltendes Vorgehen ist sinnvoll

Ein Beitrag von Herrn Prof. Mempel
Die granulomatösen Erkrankungen der Haut umfassen eine Vielzahl von verschiedenen Krankheitsbildern, die sich ursächlich in die Gruppe der infektiös bedingten und nicht infektiös bedingten (idiopathischen) Granulome unterteilen lassen. Obwohl keine einheitlichen Kriterien zur Definition existieren, wird die Gruppe der nicht infektiösen Granulome durch das Fehlen eines definierten Erregers, die nicht vorhandene Übertragbarkeit und einen krankheitstypischen histologischen Aufbau klassifiziert.
Die Entstehung und der Aufbau eines Granuloms sind komplex. Nach Opsonierung und Erkennung des auslösenden Agens spielen Antigen-präsentierende Zellen (APC) eine zentrale Rolle. Vor allem gewebsständige Makrophagen, aber auch dendritische Zellen nehmen die Erreger auf und verändern dann ihren Zellphänotyp. Typisch ist dabei die Fusion zu mehrkernigen Riesenzellen, die häufig im Zentrum eines Granuloms zu finden sind.
Bei der Auseinandersetzung der kutanen Abwehr mit proinflammatorischen Bestandteilen der Erreger kommt es dann zur Induktion verschiedener Zyto- und Chemokine, von denen TNF-α im Zentrum steht. Durch den dann erhöhten Zytokin-/Chemokingradienten wandern weitere Immunzellen in die Haut ein, hier stehen neutrophile Granulozyten und CD4-positive T-Lymphozyten im Vordergrund.
Grundsätzlich sind Entstehung und histologischer Aspekt zwischen Erreger-positiven und Erreger-negativen Granulomen nicht unterschiedlich, bei ersteren kann jedoch inzwischen fast immer ein Nachweis des auslösenden Agens (Mykobakterien, Leishmanien etc.) geführt werden [1, 2, 3, 4].
Komplizierend kommt in der Nomenklatur der Granulome eine historische Einteilung nach histologischen Kriterien hinzu, so in die tuberkuloiden, sarkoidalen, kollagenolytischen (früher nekrobiotischen), palisadenförmigen und Fremdkörper-Granulome [3, 4].
Sarkoidose der Haut
Die Sarkoidose ist eine Multi-Organ-Erkrankung mit Entwicklung lang persistierender Granulome. Bei diesen kommt es in der mittleren Dermis zu der Entstehung sogenannter „nackter“ Granulome mit wenig Infiltration von T-Zellen, der Ausbildung von sogenannten Langhans-Riesenzellen (mehrkernig aufgebaute fusionierte Makrophagen) und dem Fehlen einer zentralen Nekrosezone (im Gegensatz zu den tuberkuloiden Granulomen) [3, 4].
Die Sarkoidose ist eine relativ häufige Erkrankung, ihre Inzidenz in Deutschland wird auf etwa 12/100.000 geschätzt. Am häufigsten betroffen sind die Lungen, in unterschiedlicher Ausprägung findet man den Befall der Haut, Augen, Gelenke, des ZNS, des ossären Systems, der Tränen- und Speicheldrüsen sowie einer Vielzahl weiterer Organsysteme [5].
Die Erkrankung ist regional unterschiedlich ausgeprägt, eine hohe Prävalenz wird in Skandinavien beschrieben, niedrige Prävalenzen aus Russland berichtet. Der Erkrankungsgipfel liegt zwischen dem dritten und vierten Lebensjahrzehnt; Frauen sind häufiger betroffen als Männer.
Als prädisponierend gelten verschiedene HLA (Human Leukocyte Antigen) -Muster (HLA-B8, -DR B1, -DR B14, DR B15 und -DQ B1). Darüber hinaus sind bestimmte Verlaufsformen und Organbeteiligungsmuster ebenfalls mit verschiedenen HLA-Mustern assoziiert (HLA-DQ B1*0201 und HLA-DR B1*0301). Diskutiert wird zumindest für die akute Form die Rolle von Vimentin als Autoantigen [6].
Klinik
Klassisch wird die Sarkoidose der Haut in eine groß- und eine kleinknotige Form mit zahlreichen Sondermanifestationen eingeteilt. Pathophysiologisch erscheint eine Einteilung in eine frühe (akute) und eine späte (chronische) Form sinnvoll, auch wenn der Übergang zwischen den Verlaufsformen nicht klar definiert werden kann.
Zur akuten Form der (Haut-)Sarkoidose gehören das Erythema nodosum, die Iridozyklitis und die bihiläre Lymphknotenschwellung in der Lunge. Zusätzlich können Arthralgien (Gelenkbeschwerden) und Fieberschübe auftreten. Diese Form wird als Löfgren-Syndrom bezeichnet und zeigt einen eher entzündlichen Charakter der Erkrankung. Bei der chronischen Verlaufsform stehen dann Granulombildung und nachfolgend Fibrose, vor allem des Lungengewebes, im Vordergrund.
[accordion title=““ open1st=“0″ openAll=“0″ style=““][accordion_item title=“derma.plus Expertenwissen: Erythema nodosum„]Schmerzhafte Entzündung des Unterhautfettgewebes, die mit Knötchenbildung einhergeht. Die betroffenen Bereiche stellen sich als unscharf begrenzte, rötlich bis violette Flecken dar, die stark druckempfindlich sind.[/accordion_item][/accordion]
[accordion title=““ open1st=“0″ openAll=“0″ style=““][accordion_item title=“derma.plus Expertenwissen: Iridozyklitis„]Eine Entzündung von Iris und Ziliarkörper, die sich durch Lichtempfindlichkeit, Schmerzen und Sehstörungen äußert.[/accordion_item][/accordion]
[accordion title=““ open1st=“0″ openAll=“0″ style=““][accordion_item title=“derma.plus Expertenwissen: Fibrose„]Eine krankhafte Vermehrung von Bindegewebe, die zur Verhärtung des betroffenen Organs und letztendlich zum Funktionsverlust führt. [/accordion_item][/accordion]
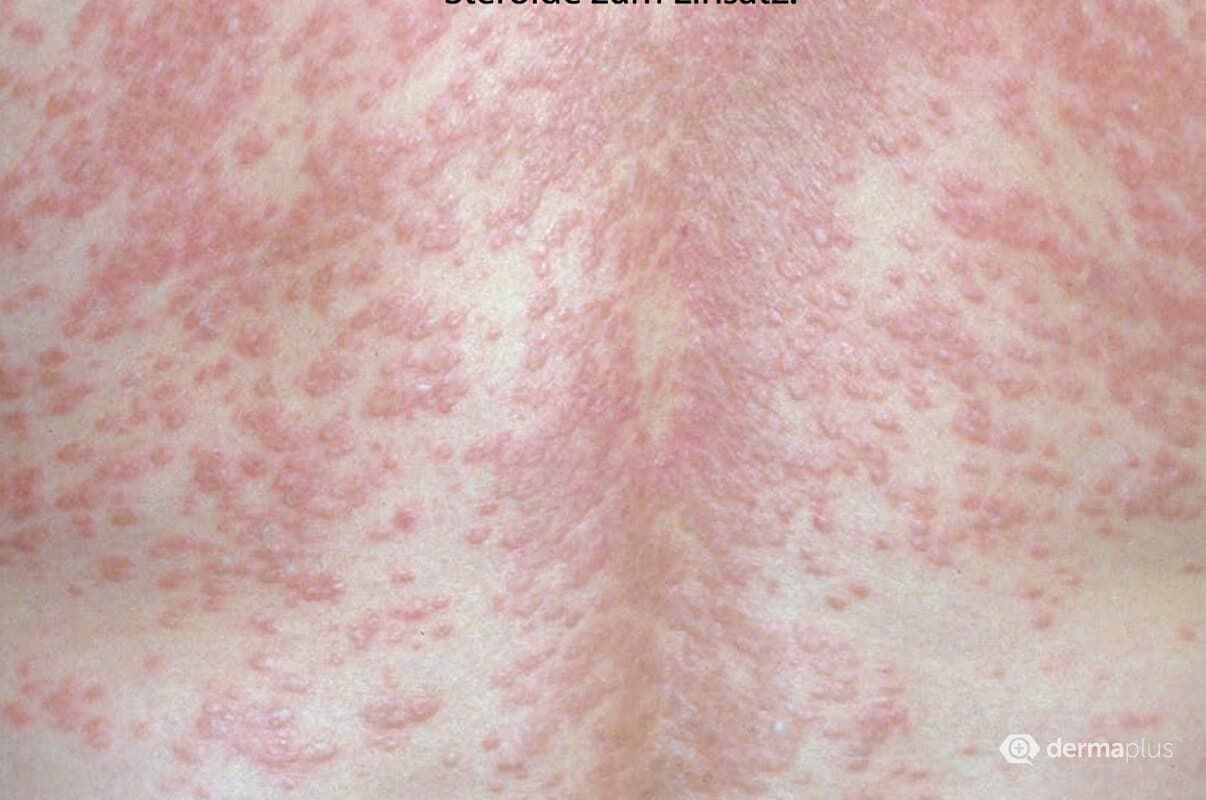
Bei der kleinknotigen Sarkoidose kommt es zum Auftreten von symptomlosen, hellrot bis livid-bläulich-roten Knötchen, die zum Teil einzeln, zum Teil gruppiert beetartig auftreten können. Es fehlt eine epidermale Beteiligung. Diagnostisch verändert sich das Infiltrat der Papeln unter Glasspateldruck zu einem gelblichen, geleeartigen Farbton. Prädilektionsstellen sind die Streckseiten der Extremitäten sowie der Rumpf (Abb. 1).
Die großknotige Verlaufsform bildet flächige, zum Teil figurierte Plaques, die einen rötlich-blauen oder rötlich-braunen Farbton aufweisen. Prädilektionsstellen sind Rumpf und Gesicht; im Gesicht besonders die Nase, die im Farbton einer Frostbeule ähnelt und so die Bezeichnung Lupus pernio geprägt hat. Sonderformen sind die zirzinäre (oder gyrierte) Sarkoidose beziehungsweise die subkutan-knotige Form (Roissy-Darier). Die Läsionen können ulzerieren (ulzerierte Sarkoidose). Eine Besonderheit ist die Narbensarkoidose, bei der sarkoide Granulome im Verlauf einer Wundheilung auftreten (Abb. 2).
Da die Sarkoidose eine potenzielle Systemerkrankung darstellt, ist die Abklärung von Organbeteiligungen unabdingbar. Am häufigsten ist die Lunge befallen. Abzuklären sind darüber hinaus eine mögliche Beteiligung der Augen (Mikulicz-Syndrom), des Nervensystems (Heerfordt-Syndrom), des Knochenapparates (Ostitis cystoides multiplex) und der Gelenke (Löfgren-Syndrom). Bei Vorliegen einer häufig gesehenen Kalziurie ist eine Nierenbeteiligung abzuklären.
Die Lungenbeteiligung tritt in vier unterschiedlichen Stadien auf, die von einer (bi-)hilären Lymphadenopathie über eine Parenchymbeteiligung bis zur Fibrose gehen kann. Man vermutet, dass bei bis zu 30 % der betroffenen Lungensarkoidosepatienten auch eine Beteiligung des Herzens, typischerweise in Form einer Reizleitungsstörung, auftritt.
Beim Löfgren-Syndrom handelt sich um eine Kombination von Erythema nodosum und/oder Arthritis oder Periarthritis der Knöchel mit bilateraler hilärer Lymphadenopathie. Häufig bestehen Fieber und Abgeschlagenheit.
Das Heerfordt-Syndrom stellt eine febrile entzündliche granulomatöse Erkrankung an Augen, Parotis und anderen Speicheldrüsen sowie dem ZNS dar. An den Augen findet man Konjunktivitis mit transparenten bräunlichen Knötchen, Keratoconjunctivitis sicca (trockenes Auge) oder Iridozyklitis mit bräunlichen Stippchen, auch Chorioiditis (Aderhautentzündung). Die Parotisschwellung (Schwellung der Ohrspeicheldrüse) ist meist doppelseitig, desgleichen die Erkrankung von Tränen-, Speichel- oder Submaxillardrüsen.
Das Mikulicz-Syndrom beschreibt ursprünglich die Kombination aus beidseitiger Parotis- und Tränendrüsenschwellung mit Siccasymptomatik. Ob es sich bei der ursprünglichen Beschreibung um eine Manifestation der Sarkoidose handelte, ist unklar; retrospektiv kommt eher ein Lymphom in Betracht.
Bei Knochenbefall bietet sich röntgenologisch das Bild der Ostitis multiplex cystoides (Jüngling-Krankheit). Dabei handelt es sich zum einen um eine trabekuläre Osteoporose innerhalb der Endphalangen (Fingerendglied), zum anderen röntgenologisch um sichtbare kreisrunde Aufhellungen [1].
Diagnostik
Neben dem klinischen Aspekt ist eine organbezogene Abklärung wichtig. Radiologisch bietet sich beim – am häufigsten auftretenden – Lungenbefall das Bild einer hilären Lymphadenopathie (Vergrößerung der Lymphknoten im Bereich des Lungenhilus), gegebenenfalls mit Parenchymbeteiligung, die bei typischem Befallsmuster im hochauflösenden CT diagnostiziert werden kann. Als Ergänzung dient der CD4-/CD8-TH-Zellenquotient in der bronchoalveolären Lavage sowie das Angiotensin-konvertierende Enzym (ACE) und der lösliche Interleukin-2-Rezeptor (IL-2R) im Serum [7].
Für den Befall der Haut ist fast immer eine bioptische Abklärung notwendig. Diese zeigt typischerweise „nackte“ Granulome (geringe Infiltration von Lymphozyten) in der mittleren bis tiefen Dermis. Wegweisend sind Epitheloidzellen, die zu mehrkernigen Riesenzellen fusionieren können (Langhans-Riesenzellen). Die zentrale Nekrose („Verkäsung“) der tuberkuloiden Granulome fehlt. Die Färbungen für Mykobakterien (z. B. Ziehl-Nelsen) sind negativ [3, 4].
Treten dermatologische Veränderung als erste Manifestation der Sarkoidose auf, ist eine Abklärung des Lungenstatus und eine symptomorientierte Untersuchung möglicher weiterer beteiligter Organsymptome sinnvoll.
Ein interessantes Phänomen bei Sarkoidose ist, dass aufgrund einer verminderten Aktivität des Immunsystems eine Anergie gegenüber Tuberkulin sowie verschiedenen anderen Antigenen wie Trichophytin, Candida albicans, Histoplasmin, Bakterienantigenen und Viren besteht [1].
Therapie
Vor Beginn einer Therapieentscheidung ist es notwendig, sich ein Bild über Ausdehnung der Erkrankung und Verlauf zu machen. Etwa 25–30% der Patienten entwickeln einen chronischen Verlauf. Dem gegenüber stehen 70–75%, bei denen eine spontane Abheilung stattfindet [5]. Stabile Befunde sind selten therapiepflichtig. Die Entscheidung zu einer Therapie hat keinen gesicherten Einfluss auf den Krankheitsverlauf.
Mittel der Wahl sind Kortikosteroide, die bei Hautbefall topisch oder intraläsional verabreicht werden können, bei Systembefall und Therapieindikation ist jedoch eine systemische Gabe notwendig. Wichtige Indikationen sind Leber-, Nieren- oder Herzbeteiligung sowie das Auftreten einer Hyperkalziämie. Die Dosierung sollte mit etwa 0,5 mg/kg KG begonnen und über einen längeren Zeitraum ausgeschlichen werden.
Alternativ kommen Chloroquin, Allopurinol, Dapson und Methotrexat zum Einsatz. Für TNF-α-Blocker (off-label) wurden sowohl krankheitsverbessernde als auch krankheitsauslösende Effekte beschrieben. Aufgrund der klinischen Ähnlichkeit mit der Tuberkulose ist bei einem Behandlungsversuch obligatorisch eine latente TBC (Quantiferon-Test) auszuschließen. Bei der akuten Form des Löfgren-Syndroms sind nicht steroidale Antirheumatika (NSAID) effektiv [1,8].
Granuloma anulare
Das Granuloma anulare ist eine auf die Haut beschränkte granulomatöse Erkrankung, bei der es zum Auftreten eines kollagenolytischen (früher „nekrobiotischen“) Granuloms in der Dermis, gelegentlich auch in der Subkutis kommt [9]. Kollagenolytisch beschreibt die atypische Anordnung von Kollagen, Muzin beziehungsweise Glykogen, die in einer Abgrenzungsreaktion von Histiozyten umgeben sind. Häfig kommt es zur typischen Aufreihung in „Palisadenstellung“ [3, 4].
Die Erkrankung tritt in verschiedenen klinischen Erscheinungsformen auf. Kinder sind häufiger von der lokalisierten Form mit Beteiligung der Akren betroffen, bei Erwachsenen dominieren disseminierte Formen. Genaue Zahlen über die Inzidenz liegen nicht vor. Eine Assoziation mit Diabetes mellitus oder einer HIV-Erkrankung wurde beschrieben, ist aber umstritten [10, 11].
Die ätiopathogenetische Ursache ist unklar. Vermutet wird eine Stimulation der Histiozyten durch Kollagenbestandteile, die dann wiederum zur Infiltration von CD4-positiven T-Zellen führt [12].
Klinik
Typischerweise treten bei den lokalisierten Formen an Hand- und Fußrücken, Streckseiten der Finger und Zehen sowie in Gelenknähe symptomlose Knötchen und Plaques auf, die sich dann in ringförmiger, zentrifugaler Anordnung vergrößern. Eine epidermale Beteiligung fehlt, der Farbton reicht von hellrot bis zu rotbraunen Läsionen. In der Wachstumsphase kommt es nicht selten zum zentralen Einsinken, sodass der namensgebende Ring entsteht (Abb. 3 & 4). Die Läsionen persistieren lange, heilen jedoch vor allem im Kindesalter häufig spontan ab. Nach zwölf Monaten sind circa 70% der Läsionen verschwunden [13].
Die Erkrankung tritt in unterschiedlichen Formen auf. Beschrieben ist das erythematöse Granuloma anulare mit sehr flachen, nur gering infiltrierten Erythemen (entzündungsbedingte Hautrötungen), die unterschiedliche Anordnungen aufweisen können. Das papulöse Erythema anulare ist die klassische Form mit ringförmigen Läsionen. Eine Sonderform stellt das perforierende Granuloma anulare dar, bei der es zu Ulzeration der Granulome kommt.
Subkutane Formen des Granuloma anulare sind klinisch schwer zu diagnostizieren und bedürfen fast immer einer histologischen Sicherung, um eine Abgrenzung gegenüber rheumatischen Knoten durchführen zu können. Im Erwachsenenalter tritt häfig die disseminierte Form des Granuloma anulare auf, bei der kleinere Papeln in flächiger Aussaat dominieren. Diese Form neigt zur chronischen Persistenz (Abb. 5).
Eine Besonderheit stellt das interstitielle (histologisch auch als „inkomplett“ beschriebene) Granuloma anulare dar, bei dem es nicht zum Aufbau eines vollständigen Granuloms kommt. Im Interstitium (Zellzwischenraum) liegen Histiozyten und Lymphozyten um schüttere Muzinbündel. Klinisch und histologisch ist die Abgrenzung zur eher bei rheumatischen Erkrankungen vorkommenden interstitiellen granulomatösen Dermatitis sehr schwierig. Ob beide Erkrankungen eigenständig sind, ist unklar (Abb. 6 & 7).
Differentialdiagnosen sind andere granulomatöse Erkrankungen wie Fremdkörpergranulome, die Sarkoidose, Necrobiosis lipoidica, Mykobakteriosen und granulomatöse Mykosen.
Histologie
Im Zentrum des Granuloms findet sich degeneriertes, in der Hämatoxylin-Eosin-Färbung blass erscheinendes Kollagenmaterial. In der Peripherie lagern sich gewebsständige Makrophagen in Palisadenformation an. Angrenzend zeigen sich lymphozytäre Infiltrate. Demgegenüber steht eine weniger gut abgegrenzte Form des Granuloma anulare, das interstitielle Granuloma anulare. Hier findet sich keine klare kollagenolytische Zone; Histiozyten und lymphozytäre Extravasate liegen in loser Anordnung zwischen kollagenen Fasern. Die Abgrenzung zur interstitiellen granulomatösen Dermatitis, die ursprünglich nur bei rheumatischen Erkrankungen beschrieben wurde, ist schwierig [3, 4].
Therapie
Bei der Entscheidung zur therapeutischen Intervention gilt es zu bedenken, dass vor allem bei Kindern und Jugendlichen ein Großteil der Hautveränderungen spontan rückläufig ist. Besteht die Indikation zur Behandlung (mechanisch/kosmetisch störend), sind lokal angewendeteSteroide Mittel der Wahl. Im Kindesalter bevorzugt in topischer Form, gegebenenfalls unter Okklusion, bei Erwachsenen auch intraläsional.
Für disseminierte Formen ist eine Vielzahl von Therapien beschrieben, die beobachteten Patientenkollektive sind jedoch meist begrenzt. Hier kommt neben einer Phototherapie (PUVA, UV-A-1) die Behandlung mit systemischen Steroiden, Fumarsäureestern, Dapson, Chloroquin und TNF-α-Inhibitoren in Betracht. Ein guter Behandlungserfolg wurde auch für Vitamin E beschrieben [14, 15].
Necrobiosis lipoidica
Der Ursprung der Necrobiosis lipoidica ist unklar. Ihr Auftreten ist mit Diabetes mellitus assoziiert, was erhöhte Zuckerwerte und damit verbunden die Glykosylierung von dermalen Proteinen als Ursache vermuten lässt. Dagegen spricht, dass nur circa 10–40% der Patienten an einem Diabetes mellitus leiden und eine Normalisierung der Blutzuckereinstellung im Rahmen einer Diabetestherapie nicht vermehrt zur Abheilung der Hautveränderungen führt. Patienten mit der seltenen genetischen Erkrankung Ataxia teleangiectatica erkranken häufiger an Necrobiosis lipoidica [1].
Klinik
Betroffen sind typischerweise die Streckseiten der Unterschenkel, wo rundliche bis kreisförmige gelb-braune Verhärtungen der Haut, häufig als tastbare Plaques, auftreten. In der Peripherie zeigt sich ein rötlich-livider Randsaum. Die Läsionen sind mit Teleangiektasien besetzt. Die Hautveränderungen treten in der Regel parallel auf, wobei der Ausprägungsgrad an den Unterschenkeln stark variieren kann. Etwa in einem Drittel der Fälle sind Ulzerationen beschrieben, die in schweren Fällen bis an den Knochen reichen können. Der Verlauf ist fast immer langwierig (Abb. 8 & 9).
[accordion title=““ open1st=“0″ openAll=“0″ style=““][accordion_item title=“derma.plus Expertenwissen: Teleangiektasien„]Mit dem bloßen Auge sichtbare, irreversible Erweiterung von kleinen Gefäßen (Kapillaren), in der Haut/Schleimhaut. [/accordion_item][/accordion]
Differentialdiagnostisch sind andere granulomatöse Erkrankungen (Sarkoidose, Granuloma anulare) sowie insbesondere die zirkumskripte Sklerodermie abzugrenzen. Die Granulomatosis disciformis chronica et progressiva (Miescher) entspricht der Necrobiosis lipoidica und stellt keine eigenständige Erkrankung dar.
Histologie
In der oberen, mittleren und tiefen Dermis, bis an das Fettgewebe heranreichend, finden sich kollagenolytische („nekrobiotische“) Kollagenbezirke; typischerweise ohne Muzinablagerungen, in denen sich die namensgebenden Lipidablagerungen nachweisen lassen. Am Rand imponieren histiozytäre lnfiltrate, zum Teil in Palisadenstellung, ferner auch Riesenzellen und Plasmazellen. Seltener zeigen sich gut abgegrenzte sarkoidale Granulome. Im Gegensatz zum Granuloma anulare weist die Necrobiosis lipoidica eine Beteiligung der gesamten Dermis auf sowie häufig ein perivaskuläres Entzündungsinfiltrat, in dem sich Plasmazellen finden [3, 4].
Therapie
Aufgrund des oft langwierigen Verlaufs ist die Therapie schwierig. Grunderkrankungen wie Diabetes mellitus sollten eingestellt werden. Mittel der Wahl ist die topische Applikation von Steroiden, mitunter als okklusiver Folienverband. Alternativ können infiltrierte Plaques mit der lokalen Injektion von Steroiden therapiert werden.
Von Vorteil ist eine milde Kompression der Unterschenkel. Durchblutungsfördernde Medikamente (ASS, Pentoxyphyllin, Heparin) wurden beschrieben. In kleinen Fallzahlen wurde von einem Ansprechen auf die photodynamische Therapie, auf Fumarsäureester, PUVA-Therapie, Thalidomid und TNF-α- Blockade berichtet [16].
Idiopathisches aseptisches Granulom des Gesichtes
Synonyme sind „Idiopathic facial aseptic granuloma (IFAG)“ und „Pyodermite froide du visage“. Dieses relativ neu beschriebene Krankheitsbild betrifft Kleinkinder, bei denen es im Bereich der Wangen zum Auftreten von Papeln oder Knoten kommt, die einem einschmelzenden Abszess ähneln. Die Hautveränderungen sind jedoch schmerzlos, nicht überwärmt, und Allgemeinsymptome wie Fieber fehlen. Die Stichinzision erbringt keinen Eiter. Histologisch finden sich mehrkernige Riesenzellen mit Ausbildung eines lymphozytären Granuloms [17].
Das idiopathische aseptische Granulom des Kindesalters heilt selbstlimitierend ab, eine oftmals veranlasste Antibiose erbringt keinen Vorteil. Abzugrenzen sind abszedierende Entzündungen der Haarwurzel, das Pilomatrixom und infektiöse Granulome wie Leishmaniose beziehungsweise Mykobakteriose. Die Erkrankung wird auch als Sonderform einer granulomatösen Rosazea im Kindesalter eingeordnet [18].